Introduction
Epilepsy in hypocalcemic patients has been reported for long period but pathophysiology underlying the hypocalcemic seizure remains to be elucidated. There have been limited reports on their semiology or EEG findings. Hypocalcemia induce a generalized seizure by changing neuroexcitability and seizure threshold.1,2 In addition, atypical absence, atonic, and brief focal (partial) seizure are also reported in these patients.1,3 Previous EEG findings include slowing of background rhythm, diffuse increase in slow wave activities with frontal preponderance, increased fast activity and multifocal spikes.1,4
Up to 20–25 % of patients with acute hypocalcemia present epileptic seizure and up to 70% of chronic hypocalcemia patients are associated with epilepsy.1 Common etiologies for chronic hypocalcemia are vitamin D deficiency and parathyroid hormone (PTH) dysfunction such as hypoparathyroidism (low PTH level) or pseudohypoparathyroidism (ineffective peripheral PTH receptor).2
In chronic parathyroid dysfunction, as many as fifty percent of the patients have intracranial calcification in deep GM and subcortical WM of their brain and some of them are also epilepsy patients.3 However, cortical calcification has not been demonstrated in these patients. In mid 1900s, researchers reported calcification in frontal or occipital cortices in hypocalcemic patients but their figures illustrated calcification in subcortical fibers rather than cortical GM with skull radiographs and one early computed tomography (CT) scan with limited resolution.3 What the researchers meant by cortex seemed cerebral lobes including WM fibers and the point of papers was that calcification outside BG also exists. There have been no studies with modern CT or MRI reporting cortical calcification in chronic hypocalcemia.
The relationship between cortical calcification and epileptic seizure had been demonstrated in other brain disease such as neurocysticercosis or Sturge-Weber Syndrome (SWS).5 Here, we report a focal seizure patient with pseudohypoparathyroidism, who had intracranial calcification in cortical as well as subcortical areas.
Case
36 year old male came to our clinic for evaluation of seizure episodes which started two years ago. His parents witnessed him stop talking in the conversation and staring blank 3 to 4 times a day. Initially his symptom was subtle to be noticed but later his symptom became concrete to be recognized by the patient himself. The frequency of complex partial seizures was 3 to 4 times a day. He was given levetiracetam 500 mg a day for two weeks which only had partial response. We admitted him for epilepsy evaluation. Previous his medical records included a pre-term born and a laser ablation of bilateral cataracts at the age of thirty three years old. He had no family history of any endocrine or neurologic diseases. Clinical examination did not reveal any physical abnormalities associated with Albright-hereditary osteodystrophy or rickets. The neuropsychological tests revealed mild learning and memory impairment although his score of full scale IQ was 105.
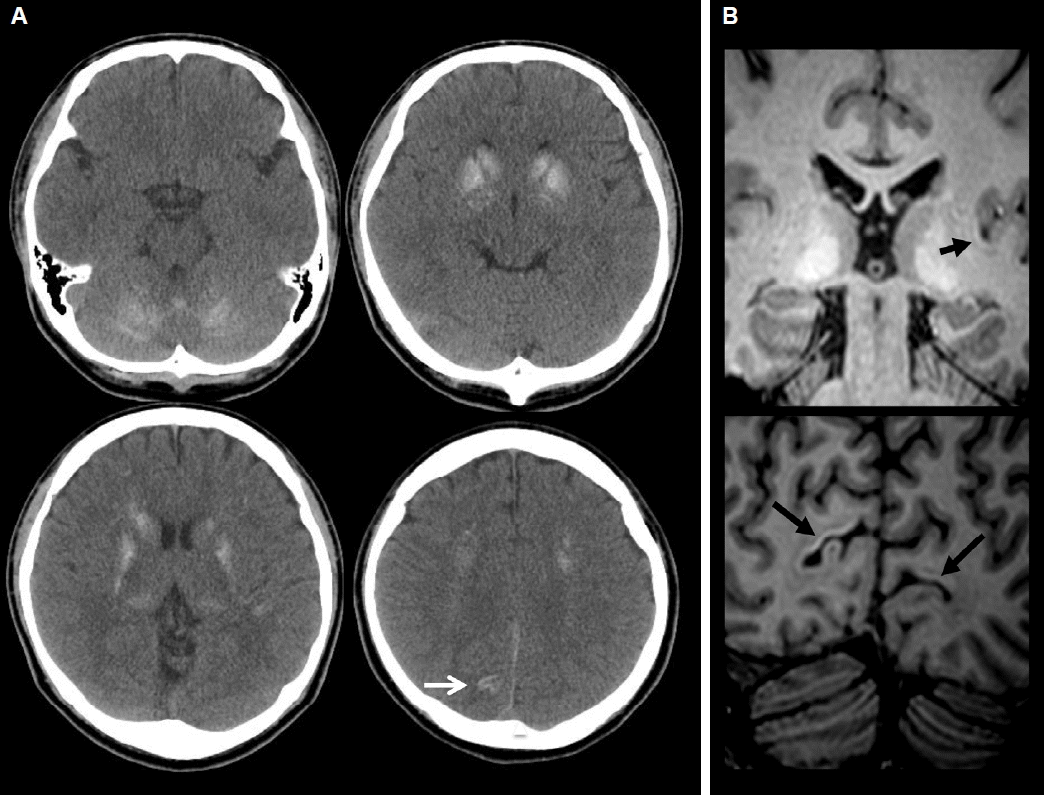
His blood lab showed low calcium level 5 mg/dL (normal range: 8.0–10.4 mg/dL) and high parathyroid hormone 114 pg/mL (normal range: 15–68 pg/mL) and phosphorous level 6.3 mg/dL (normal range: 2.5–4.5 mg/dL). Additional parameters measured are listed in Table 1. CT (Fig. 1A) and T1 magnetic resonance imaging (MRI) (Fig. 1B) scan of his brain revealed bilaterally symmetrical calcification in basal ganglia (BG), thalami, cerebellar hemispheres and subcortical WM of bilateral frontoparietal lobes. In addition, calcifications along left insula and bilateral occipital cortical ribbon were present.
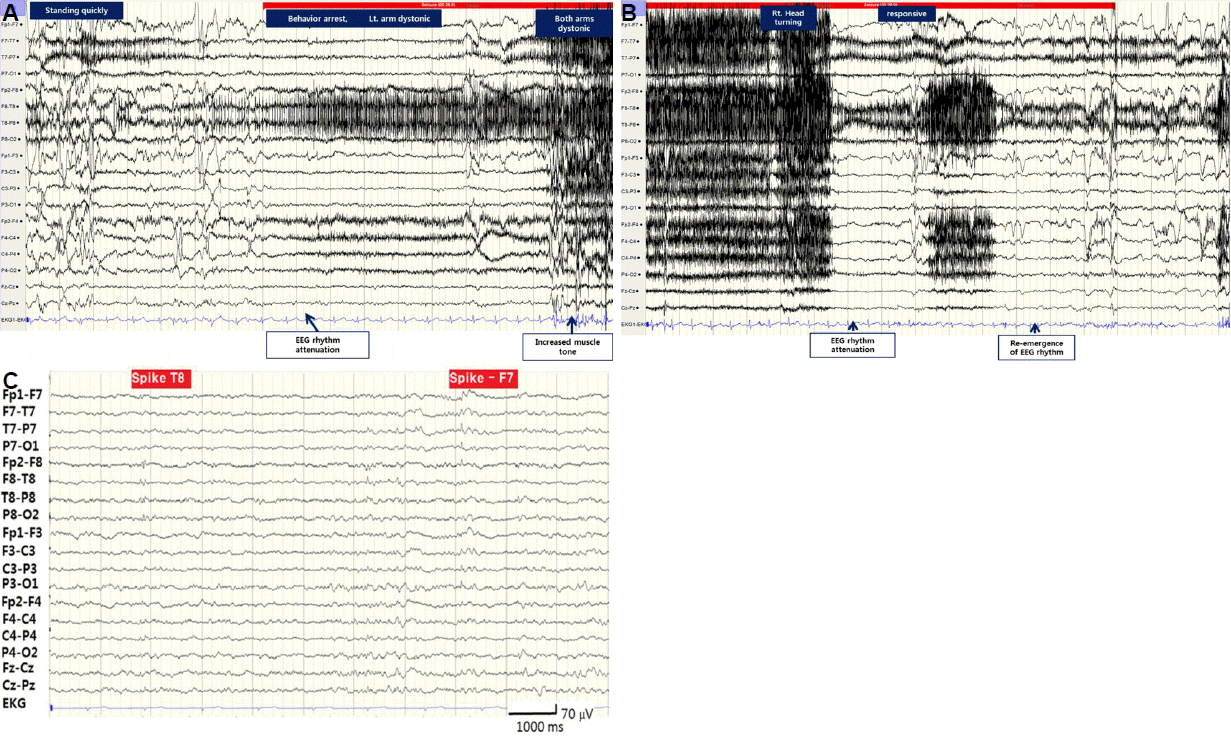
During video-EEG monitoring, he had 6 seizures which usually started when he made a sudden movement. He felt abnormal sensation of right leg followed by behavior arrest, unresponsiveness and dystonia of left arm. Brief myoclonic jerk then followed and his head turned toward right. The ictal EEG changes were initiated (Fig. 2A, B) with polymorphic delta (2–3 Hz) wave at right hemisphere, which showed maximum activity in right temporal region. Background attenuation followed then muscle artifact appeared as the patient’s left arm became dystonic. Semirhythmic delta wave reappeared at right hemisphere, the rhythmic delta activity continued to rhythmic theta wave and became beta wave at the end of the ictal rhythm. The interictal EEG (Fig. 2C) showed multifocal spikes shown mainly over left frontotemporal (51.1%) over right frontotemporal hemisphere (27.7%).
After monitoring, levetiracetam 500 mg per day was restarted with supplement of calcium carbonate 1,500 mg and calcitriol 0.25 mcg per day. He became seizure free while his calcium and phosphorous levels almost normalized at the three months follow up.
Discussion
In this case, we described a focal epilepsy patient with pseudohypoparathyroidism and intracranial calcification. Although hypocalcemia alone can cause epileptic seizure, focal metabolic seizure is rare. Thus we hypothesized that his unusual distribution of calcification in cortical ribbon were related to the epileptogenicity of his partial seizure.
By EEG-video monitoring, we attempted to localize the epileptogenic focus of his seizure. Interictal spikes were multifocal in nature and predominant over left (72.3%) versus right (27.7%) frontotemporal regions. This could be result of the low serum calcium level but also of the cortical calcium deposit as left temporal spikes were most frequently seen correlated with left temporal calcification. During the ictal phase, the seizure started with right somatosensory aura followed by left limb dystonic posturing and right head turning and staring. The somatosensory aura in right leg suggests the ictal symptomatogenic areas could be located more cortically than subcortically. Somatosensory aura is known to arise mostly from contralateral postcentral gyrus but also sometimes from ipsilateral secondary somatosensory area (SSA) or temporal cortices.6,7 According to previously studied semiology localization, succeeding left limb dystonic posturing accompanied by right head turning suggest the ictal focus is in the right hemisphere propagated to the right BG.6 Thus, it is likely that his aura originated from right hemisphere, either SSA or temporal area, and spread to ipsilateral BG. The ictal EEG findings of delta to theta waves with maximal activity at the right hemisphere, especially right temporal lobe, also support this assumption. Unfortunately, his CT and T1 MRI showed calcification most clearly in left temporal and bilateral occipital cortices, not right temporal or parietal cortices. Intracranial calcifications with calcium concentration of 30% or less will cause T1 hyperintensity but at higher concentration, the intensity of the signal diminishes.8 Gradient echo protocol was also done but thicker sections and blooming artifact at basal cerebral surface made it harder to visualize calcification than T1 MR or CT imaging. Susceptibility Weighted Imaging (SWI), relatively recent MR protocol, has been recognized as a superior tool in visualization of calcification and SWI protocol was employed to demonstrate the calcified cortical lesion in other epileptic disease. For example, Sturge-Weber Syndrome patients with severely calcified cortex demonstrated by SWI, suffered from a higher seizure burden (a measure of seizure frequency and epilepsy duration) and had a trend for earlier seizure onset.5 Unfortunately we did not include SWI protocol when we took our patient’s MRI.
Whether intracranial calcification in other deep structure, especially BG, contributes to the epileptogenicity of these hypocalcemic epilepsy patients is unknown. Since epilepsy is known to be cortical origin, BG calcification sparing cortices has been disregarded in understanding epileptogenicity of chronic hypocalcema. However, there are studies which demonstrated the role of BG in epilepsy propagation and modulation. Some modeled BG as a seizure propagation pathway by injection of different NMDA antagonist into the substantia nigra pars reticulate suppressing both behavioral and EEG expression of epileptic seizures.9–11 Others suggested BG as a remote inhibitory circuit of epileptic seizure.12,13 Calcium deposit in BG may disrupt an inhibitory circuit and make patients with hypocamesia vulnerable to epileptic seizure. Functional imaging such as Single-photon emission computed tomography (SPECT) or positron emission tomography (PET) might be able to demonstrate functional compromise of BG by the mineral deposit.
Despite the apparent absence of familial history and without a confirmatory genetic or molecular test, the combination of biochemical abnormality, neuroimaging finding, clinical course (early cataracts, epilepsy, and treatment response) assured us the diagnosis of pseudohypoparathyroidism as the etiology for his chronic hypocalcemia. Hypocalcemia and hyperphosphetemia with high level of PTH and low level of 25-hydroxyvitamin D can be also seen in secondary hyperparathyroidism due to vitamin D deficiency. Although, vitamin D deficiency can cause hypocalcemic seizures in infants and young children1,14, adult with such severe hypocalcemia due to vitamin D deficiency and consequently epileptic seizure or intracranial calcification is very rare. Furthermore, at three months follow up, his PTH level increased regardless of calcitriol supplement. If his hypocalcemia and hyperparathyroidism are secondary due to vitamin D deficiency, calcitriol will increase serum calcium level and consequently decrease PTH level unlike his case.
In summary, we found that in addition to classical subcortical location, cortical calcification can be seen in chronic hypocalcemia due to pseudohypothyroidism. The electroclinical finding showed that the patient’s seizure arise from right temporal lobe, although the cortical calcification was most clearly demonstrated in left insular and bilateral occipital cortices. Thus, we were not able to demonstrate that these cortical deposits are directly related to epileptogenicity. However, important observations were made. Multifocal interictal spikes were seen dominantly seen over left temporal area where the mineral deposit was clearly seen. Also, during ictal phase, EEG change was mainly initiated in unilateral (right) hemisphere suggesting focal metabolic seizure originate from unilateral hemisphere as seen in other focal epilepsy. His semiology suggests possible seizure propagation to subcortical structure especially BG. Therefore, subcortical calcification commonly seen in chronic hypocalcemia patients might contribute to epilepsy propagation as well. Lastly, even in focal epilepsy patients, neuronal hyperexcitability theory is important as we noted that his seizures which initially did not respond to conventional antiepileptic drug became quickly controlled with calcium and calcitriol supplement. This could be attributed to the stability of neuronal cells by correction of hypocalcemia.
Our limitation was that we did not include more sophisticated imaging protocols like SWI in visualization of cortical calcification in our patient. Visualization of intracranial calcification in chronic hypocalcemia patients by neuroimaging will enable us to understand the nature of hypocalcemic epilepsy better and this could be a possible measure of seizure burden and prognosis in these patients.