Introduction
Benign epilepsy with centro-temporal spikes (BECTS) is the most common type of focal epilepsy in children. It is known to be age-dependent, presumably genetic, and mainly occurs at developmentally critical ages. Age of onset ranges from one to 14 years and the prevalence is about 15–20% in children younger than 15 years of age.1,2 Generally, BECTS is characterized by infrequent focal sensorimotor seizures in the face during sleep, which may secondarily generalize, along with spike-wave discharges, reflecting nonlesional cortical excitability from rolandic regions. The prognosis is usually considered to be excellent. Over the past years, however, some investigators have questioned whether BECTS is indeed benign, considering the variety of different presentations associated with the disorder. It is not uncommon for BECTS to be associated with neuropsychological deficits, such as linguistic, cognitive, and behavioral impairment.3–7 In particular, reading difficulties and speech/language disorders are more common in children with BECTS than in healthy controls.8 Various neuropsychological deficits seem to be very dependent on the sleep-wake cycle and spike index, as well as the predominant localization of epileptiform discharges.9–15 Furthermore, the frequency of epileptiform discharges is closely related not only to the degree of neuropsychological deficits, but also to an atypical evolution of BECTS. The atypical evolution of BECTS can result in atypical benign childhood focal epilepsy (ABCFE), status epilepticus of BECTS (SEBECTS), Landau-Kleffner syndrome (LKS), and epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS), which are considered as different entities, but are part of a single spectrum of disorders.16 Genetic predisposition with complex modes of inheritance has long been advocated for this wide range of epileptic syndromes, even though unraveling the pathogenic mechanisms that underlie these conditions remains a challenge. Considering the various possible developmental trajectories of the disorder, future research needs to work on evidence-based categorization of BECTS and related epileptic syndromes.
Atypical features and atypical evolution of benign childhood epilepsy with centro-temporal spikes
Typical BECTS, known as BECTS pure, is an age-dependent, presumably genetic, male predominant, focal epilepsy syndrome in children. The cardinal features typically consist of single or infrequent, brief hemifacial sensory motor seizures, usually during sleep. These seizures are often associated with oropharyngolaryngeal symptoms, such as making strange noises, speech arrest, and salivation. In about half of children with BECTS, the seizures evolve into generalized tonic-clonic seizures. The interictal electroencephalogram (EEG) is characterized by high-voltage focal or multi-focal spikes, mainly in rolandic regions, showing a tendency to spread to adjacent regions. This neural activity is common during sleep. There are no, or very few, linguistic or neuropsychiatric deficits. Seizures and EEG features usually resolve within a few years of onset, and typically before the age of 15 or 16 years.
Atypical features in BECTS comprise many clinical and electro-physiological findings. The seizures only occur in the daytime, and Todd’s paralysis can be prolonged or even present as status epilepticus. The EEG data can show an atypical morphology of epileptiform discharges, an abnormal background, unusual location, and focal or bilateral synchronous discharges, including the 3 Hz spike and wave complexes.16–21 The atypical features seem to be related to the onset of the seizures.22–25 In addition, previous studies have shown that many children with atypical features are likely to have linguistic, learning, or behavioral difficulties.15–17,20,25–28 The common comorbidities are listed in Table 1.
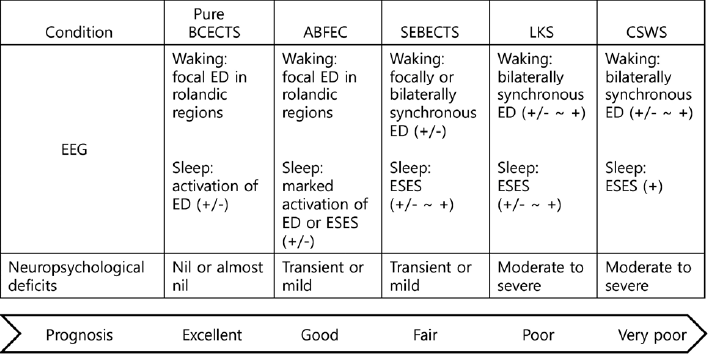
The atypical evolution of the disorder seems to be closely related to the sleep-wake cycle and the frequency of epileptiform discharges. Individuals with an atypical evolution tend to have moderate to severe linguistic, cognitive, or neuropsychological deficits, which can be permanent. The EEG data usually show a marked increase in abundance and a bilateral synchronization of epileptiform discharges in the rolandic area, or otherwise ripples superimposed on rolandic spikes, which may become continuous during NREM sleep.10,11,16,24,29 Electrical status epilepticus during slow-wave sleep (ESES) is characterized by paroxysmal or strong activation of diffuse or bilaterally synchronous spikes and waves at about 1–3.5 Hz during NREM sleep (Fig. 1). Sometimes, BECTS can evolve into ABFEC, SEBECTS, LKS, and CSWS. The degree of the neuropsychological deficits and the prognosis of each condition are illustrated in Fig. 2.
Over the past years, these conditions have been considered as different disease entities but part of a single spectrum of disorders. Genetic predisposition has long been advocated for this wide range of syndromes. Several studies have attempted to determine the association between atypical rolandic epilepsy and known genes. Although associations remain speculative, putative genes include fork head box protein P2 (FOXP2), which has been related to speech and language development, and sushi repeat-containing protein, X-linked 2 (SRPX2), which is involved in conditions of the rolandic or Sylvian areas, such as speech and language disorders, cognitive difficulties, and neuronal migration disorders. A third relevant gene is elongation factor protein 4 (ELP4), which is associated with language impairment, autism spectrum disorder, and mental retardation. Further relevant genes include the N-methyl-D-aspartate (NMDA) receptor subunit-encoding genes (GRIN2A and GRIN2B), linked to speech and language, cognitive impairment, and behavioral difficulties; the proline-rich transmembrane protein 2 (PRRT2) gene contributing to a wide spectrum of neurological diseases, ranging from mental retardation to paroxysmal neurological conditions such as paroxysmal kinesigenic dyskinesia (PKD), epilepsy and migraine; and the gamma-aminobutyric acid type A receptor subunit genes (GABAA–R and GABRG2), which have been associated with various neuropsychiatric conditions.30–40 Very recent studies showed evidence that children with typical or atypical features share known epilepsy genes, including GRIN2A and PRRT2, which can considered as different clinical phenotypes of a single pathological entity situated at the crossroads of speech and language disorders, cognitive disorders, and epilepsy.41,42
Atypical benign focal epilepsy of childhood/Pseudo-Lennox syndrome
ABFEC is a form of idiopathic focal epilepsy characterized by normal development prior to the onset of seizures and multiple seizure types. Beside the typical sensorimotor seizures of BECTS, various other types of seizures may also occur, such as secondarily generalized tonic-clonic seizures, atypical absence, myoclonic seizures, atonic seizures, and negative myoclonus at a later stage. Individuals with ABFEC are more likely to have neurocognitive, speech and language, or behavioral deficits. The waking EEG usually resembles that of BECTS, but the sleeping EEG shows the ESES pattern. The electro-clinical state is clearly related to an individual’s neuropsychological deficits. The associated seizures tend to be resistant to many anti-epileptic agents; however, they usually disappear before adolescence and the neuropsychological deficits recede with the remission of ESES.27,43,44 Pseudo-Lennox syndrome is a more severe form of ABFEC with respect to clinical features, seizure outcome, and neuro-psychiatric symptoms. Some patients remain mentally retarded even after the remission of the seizures.45–47
Status epilepticus of BECTS
SEBECTS refers to status epilepticus that can be convulsive or non-convulsive, and either generalized or focal. Besides hemifacial contraction, the seizures may present with dysfunctions of the lip, tongue, and pharynx, including speech arrest, dysarthria, excessive drooling, oromotor dyspraxia, and swallowing difficulties.17,27,46,48 The interictal EEG usually shows focally or bilaterally synchronous sharp waves or sharp and slow wave complexes with predominance in the rolandic area. The epileptiform discharges may become continuous when the individual falls asleep. Ultimate neurocognitive outcome appears good when the disorder is appropriately treated.
Landau-Kleffner syndrome
LKS, known as acquired epileptic aphasia, is an epileptic encephalopathy characterized by various types of seizures, insidious or sudden onset acquired aphasia with verbal auditory agnosia, and neuro-cognitive regression that is associated with distinctive EEG abnormalities. Incidence of LKS in children aged 5–14 years is about one in a million and prevalence in children aged 5–19 years is one in about 300,000–410,000 in Japan.49 LKS is considered as one of the rarest but most severe genetic childhood focal epilepsies. Alongside other putative genes, mutations in the GRIN2A gene (encoding the N-methyl-D-aspartate glutamate receptor alpha2 subunit, GluN2A) are thought to be related to this condition.41,50 Besides focal motor seizures, children with LKS may have generalized tonic-clonic seizures, atypical absence, atonic, and other type of seizures. The majority of children with LKS present with expressive aphasia, and more than half of these children have problems with receptive language, auditory processing, auditory working memory, and verbal memory, as well as learning difficulties and attentional and behavioral problems.51 Benign EEG patterns, like focal epileptiform discharges, may evolve into ESES during sleep in about half of children with LKS.52 A previous study showed that more than 80% of children with LKS had bilateral epileptiform discharges in the perisylvian cortex, an area of the cerebral cortex associated with speech, language, and auditory processing; and about 20% demonstrated a unilateral perisylvian hotspot that triggered secondary bilateral synchronization.53 The degree of neurocognitive deterioration may correlate with the site and spread of the epileptiform discharges. However, further studies are necessary to elucidate whether these bioelectrical abnormalities play a causal role in children with LKS. Adequate and early medical intervention may avoid language and cognitive impairments. Long-term outcome for LKS is not poor, but seizures and EEG abnormalities do not always disappear. Most children with ESES experience permanent language or cognitive impairment, depending on how well their seizures and EEG patterns respond to treatment. Overall, individuals with LKS report a poor quality of life, mainly due to language or cognitive difficulties.54
Acquired opercular epilepsy with oromotor dysfunction is a condition that is difficult to categorize because the clinical features look similar to SEBECTS and LKS. Acquired opercular epilepsy with oromotor dysfunction presents with mixed types of seizures and prolonged episodes of dysarthria, a gradual decrease in verbal output, oromotor dysfunction, and deterioration in cognitive function over several weeks to years. The EEG shows remarkably activated sharp, or sharp-slow discharges, or ESES patterns on rare occasions during NREM sleep. Magnetic resonance imaging of the brain is usually normal.
Epileptic encephalopathy with continuous spike-and-waves during slow-wave sleep
CSWS or ESES is an age-related, epileptic encephalopathy that presents at the extreme end of atypical BECTS. It is typically characterized by various types of seizures, CSWS or ESES shown on EEG during NREM sleep, and global regression of linguistic, cognitive function, and behavior. The onset of seizures varies, but the seizures tend to peak at about five years of age, before evolving into epileptic encephalopathy with ESES within 1–2 years.55 At first, the seizures may be simple focal motor, complex focal, absence, or myoclonic, and usually occur at night. They can last for more than 30 minutes. The EEG usually shows multifocal epileptiform discharges or bilaterally synchronous sharp or spike-wave discharges. Subsequently, the seizures become frequent and predominantly nocturnal. Various types of seizures then emerge. They include hemiconvulsive, generalized tonic-clonic seizures (GTCS), absence seizures, drop attacks, and convulsive or non-convulsive status epilepticus. The EEG starts showing diffuse paroxysms and a continuous pattern of ESES, which eventually occupies at least 85% of NREM sleep.27 Linguistic, neurocognitive decline, and neuropsychiatric features, such as autism, are commonly associated with this condition; the development of these features is largely dependent on the EEG pattern, including the location and abundance of epileptiform discharges. However, the pathophysiological mechanisms are still unclear. It appears that the longer the duration of ESES, the poorer the outcome is.25,56–63 Children with CSWS show differences in the degree and type of neurocognitive regression, the features of seizures, and EEG abnormalities. Therefore, it is not appropriate to generalize about this type of epilepsy. Several treatment regimens for atypical BECTS, especially LKS or ESES, have been used to improve cognitive outcome, though these are not evidence-based. Treatment options include benzodiazepines, sulthiame, new antiepileptic drugs such as lamotrigine, topiramate, levetiracetam, a ketogenic diet, steroid or methylprednisolone pulse therapy, intravenous immunoglobulin, and even surgical interventions.41,64,65
In conclusion, BECTS is the most common, presumably genetic, type of focal epilepsy in children. It can develop in various ways, leading to mild or severe neuropsychological deficits, including linguistic, cognitive, and behavioral impairment. The seizures appear to depend on the sleep cycle, and the predominant localization and abundance of epileptiform discharges. A complex interplay between the processes of brain maturation and the effects of specific genes seems to contribute to a variety of different childhood epileptic syndromes. BECTS, atypical BECTS, SEBECTS, LKS, and CSWS can all be considered different entities that are nonetheless part of a single spectrum of disorders. However, much larger cohort studies are needed using comprehensive evaluation including electro-clinical tests and whole or targeted exome sequencing, to obtain a clearer picture of what might be directly causing, or contributing to the diverse phenotypes of this spectrum of disorders.