Introduction
Lenalidomide is a derivative of the chemotherapy drug thalidomide and is mainly used for treatment of myelodysplastic syndromes.1 This medication seems to be more potent than thalidomide and achieves therapeutic responses at lower doses in both visceral and nervous system disorders.2,3 The adverse effect profile of these medication are also different. For instance, thalidomide can result in peripheral neuropathy by its cumulative dose, whereas lenalidomide rarely exhibits such side effect.2,4 Lenalidomide, when orally administrated, can penetrate the cerebrospinal fluid and be used for the central nervous system (CNS) malignancies in children.5 Neuroprotective effects of lenalidomide have also been shown in preclinical studies on neurodegenerative diseases such as amyotrophic lateral sclerosis6 and Parkinson’s disease.7 Accumulating evidence indicate an anticonvulsive property for thalidomide in different animal models of seizure such as pentylenetetrazole (PTZ)-induced clonic seizures or amygdala kindling in rodents.8–10 This raises the possibility that thalidomide derivates such as lenalidomide may also exert similar effects on seizure threshold. However, this issue has not been investigated so far.
Lenalidomide interplays with many neurotransmitters in the biological system, among which nitric oxide (NO) is an important example.11 NO has been shown to mediate both the peripheral and central actions of lenalidomide.12,13 The role of NO in the CNS function, especially in modulating seizure threshold, has been well documented in the literature.14–17 N-methyl D-aspartic acid (NMDA) receptors (NMDARs) are identified as excitatory synaptic transmission and play pivotal roles in the seizure pathogenesis, hypoxic brain damage, and other CNS diseases.18 Additionally, the NMDAR-mediated NO signaling contributes to pathogenesis of seizures and other cortical brain dysfunctions.19–21 We also recently demonstrated a role for NO pathway as an underlying mechanism in the anticonvulsive effects of thalidomide in PTZ-induced clonic seizures in mice.10,22 Therefore, the objective of the present study was to assess the effects of lenalidomide on PTZ-induced clonic seizure threshold in mice and investigate possible involvement of the nitrergic and glutaminergic pathways in this effect of lenalidomide.
Methods
Chemicals
Chemicals that were used in this study included PTZ, L-N G-nitroarginine methyl ester (L-NAME; a non-specific NO synthase inhibitor), 7-nitroindazole (7-NI; a selective neuronal NO synthase [nNOS] inhibitor), aminoguanidine (AG; a selective inducible NO synthase [iNOS] inhibitor), D-Serine (an agonist of NMDAR), and MK-801 (a selective NMDAR antagonist). All drugs were purchased from Sigma (St. Louis, MO, USA). All chemicals, except for PTZ 0.5% (which was administered intravenously), were intraperitoneally (i.p.) injected. 7-NI and lenalidomide were suspended in dimethyl sulfoxide (DMSO) 1%, while other chemicals were dissolved in normal saline.
Animals
Male NMRI mice weighing 26±3 g were purchased from Department of Pharmacology, Tehran University of Medical Sciences. Animals were kept under 12-h light/12-h dark cycle in a temperature-controlled room (22±2°C) with humidity of 55±2%. Our experiment was performed according to the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). Also, the experiment protocol has been approved by the local Ethical Committee. Eight animals were used in each group, and each animal was used only once.
Seizure induction by pentylenetetrazole
To assess the threshold of clonic seizures, a dental needle (30-gauge) was inserted into the lateral vein of mice’s tail and fixed by adhesive tape; thereafter, mouse was allowed to walk freely. Meanwhile, an infusion pump pushed the PTZ solution (0.5%) into the tail vein at a constant speed of 1 mL/min and continued until forelimb clonus followed by full clonus of the body (general clonus). The seizure threshold was calculated in mg/kg PTZ based on the time needed to appearance of the general clonus of limbs, the body weight of the animal, and the rate of infusion and concentration of PTZ in the infusate:22–25 the seizure threshold = [infusion duration (min) × infusion rate (mL/min) × PTZ concentration (mg/mL) × 1,000] / [weight of mouse (g)].
Experiments
Lenalidomide at different doses (5, 10, 20, and 50 mg/kg, i.p.) was administrated 60 minutes before PTZ-induced clonic seizure threshold assessment. The effective dose of lenalidomide (10 mg/kg, i.p.) were used for subsequent experiments. Mice were injected the equal volume of the vehicle (1% aqueous solution of DMSO) in all experiments and considered as control group. Regarding the assessment of the time course of lenalidomide, effective dose of lenalidomide (10 mg/kg, i.p.) was administered 30, 60, or 120 minutes prior PTZ. To assess the role of NO pathway, mice in separate groups received an acute administration of L-NAME (10 mg/kg), 7-NI (30 mg/kg), and AG (100 mg/kg) 15 minutes before vehicle and lenalidomide (10 mg/kg) administration or 75 minutes before PTZ administration. A single dose of MK-801 (0.05 mg/kg, i.p.) and D-serine (30 mg/kg, i.p.) was administered 15 minutes before vehicle and lenalidomide administration or 75 minutes before the administration of PTZ. This study was performed to investigate the role of NMDA receptors in the lenalidomide effects on the PTZ-induced clonic seizure threshold. The dose and time line for administration of these drugs were based on a pilot study and our previous experiments.10,17
Statistical analysis
Data were expressed as mean±standard deviation. In order to confirm the normal distribution of the data, we used homogeneity of variance test. Data were analyzed by one-way analysis of variance (ANOVA) followed by post hoc Tukey's test and p<0.05 was considered statistically significant.
Results
Effect of lenalidomide and its time courses on seizure threshold
Figure 1A illustrates the effect of the different doses of lenalidomide (5, 10, 20, and 50 mg/kg, i.p.) on the clonic seizure threshold induced by PTZ. Analyses revealed a significant anticonvulsive effect of lenalidomide at doses of 10 mg/kg (p<0.001) and 20 mg/kg (p<0.01) in comparison with control group. Figure 1B depicts the effect of time course on the anticonvulsive activity of lenalidomide (10 mg/kg, i.p.). Lenalidomide did not change the seizure thresholds when it administered 30 minutes before the PTZ infusion, but it had a maximum anticonvulsive property 60 minutes after injection (p<0.001, compared with the control group).
Effect of L-NAME on the anticonvulsive effects of lenalidomide
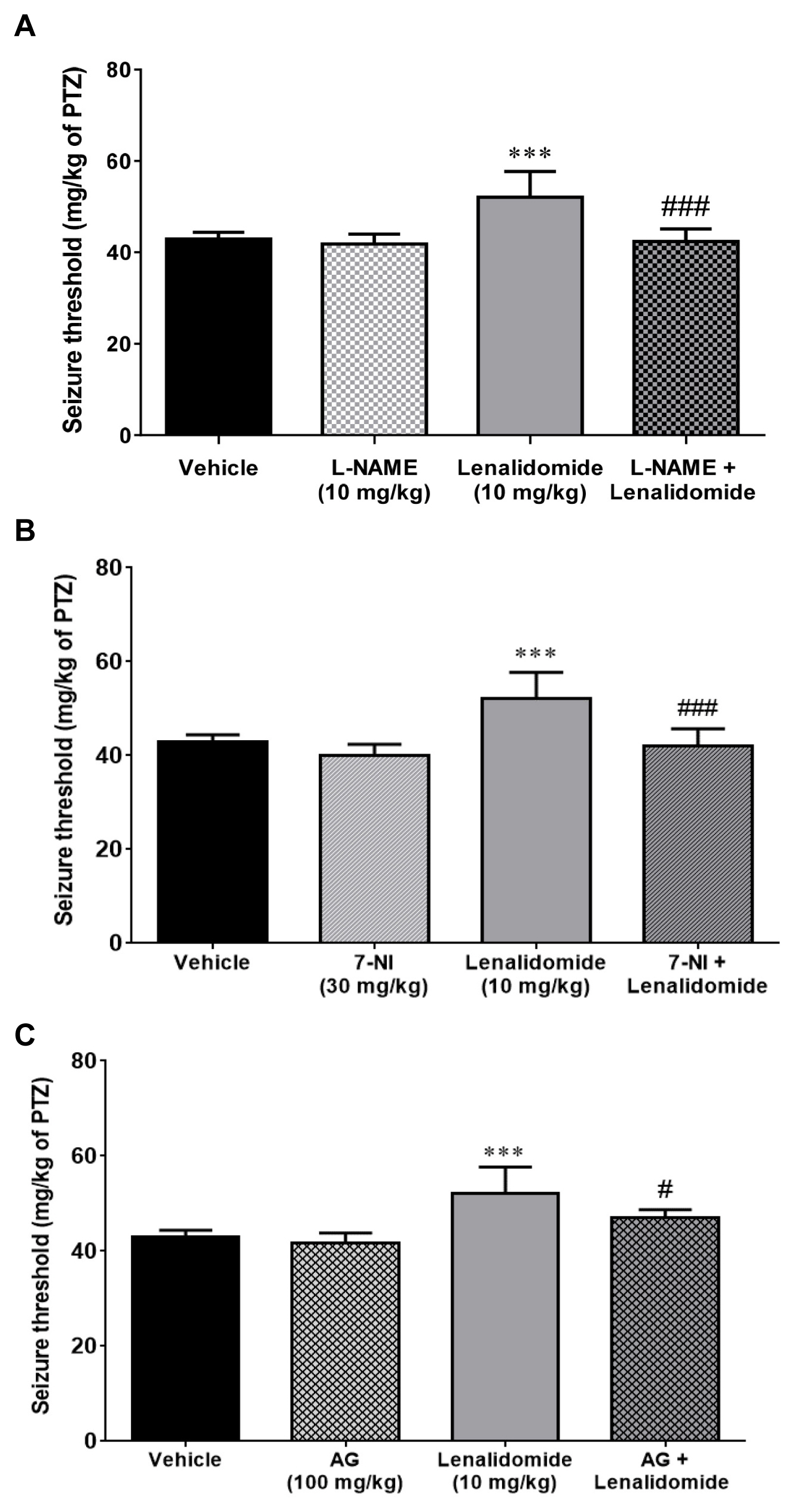
Figure 2A demonstrates the effect of L-NAME injection, alone and 15 minutes before vehicle or lenalidomide (10 mg/kg, i.p), on the PTZ-induced clonic seizure threshold. Although L-NAME (10 mg/kg, i.p.) per se did not alter seizure threshold compared with the control group (p>0.05), it prevented anticonvulsive effects of lenalidomide on seizure threshold (p<0.001).
Effects of 7-NI on the anticonvulsive effects of lenalidomide
As shown in Figure 2B, the non-effective dose of 7-NI (30 mg/kg, i.p) did not affect the threshold of seizure when injected alone compared to the control group (p>0.05). However, pretreatment with the same dose of 7-NI, 15 minutes before the administration of lenalidomide, markedly reduced the anticonvulsive influence of lenalidomide (10 mg/kg, i.p; p<0.001).
Effect of AG on the anticonvulsive effects of lenalidomide
As represented in Figure 2C, administration of AG alone (100 mg/kg, i.p.) did not have any significant effect on seizure threshold compared to the control group. Pre-treatment with AG (100 mg/kg, i.p) 15 minutes before the administration of effective dose (10 mg/kg) of lenalidomide significantly (p<0.05) reversed the anticonvulsive effect of lenalidomide in comparison to the lenalidomide-treated group.
Effects of NMDAR agonist (D-serine) and antagonist (MK-801) on the anticonvulsive effects of lenalidomide
Figure 3 illustrates effects of D-serine and MK-801 on the PTZ-induced clonic seizure thresholds in mice. Administration of the non-effective dose of D-serine (30 mg/kg, i.p) and MK-801 (0.01 mg/kg, i.p) per se was not associated with any significant alteration in the seizure threshold compared to control group. In addition, co-administration of D-serine (30 mg/kg, i.p) with lenalidomide did not change the effects of lenalidomide on the PTZ-induced clonic seizure thresholds compared to the lenalidomide (10 mg/kg, i.p) group (p<0.05). In contrast, combination of MK-801 (0.01 mg/kg) with lenalidomide (10 mg/kg, i.p) markedly decreased anticonvulsive effects of lenalidomide (10 mg/kg, i.p).
Discussion
The present study, for the first time, demonstrated that lenalidomide can markedly increase the PTZ-induced clonic seizure thresholds in mice. We also evaluated the possible involvement of both NMDAR and NO signaling pathways in the anticonvulsive effects of lenalidomide. To assess involvement of NO signaling, we used different NOS inhibitors including the non-selective NOS inhibitor L-NAME, the selective nNOS inhibitor 7-NI, and the selective iNOS inhibitor AG and evaluated their effects on anticonvulsive effects of lenalidomide. Pre-treatment with a per se non-effective dose of each of these NOS inhibitors in separate set of experiments significantly inhibited or attenuated the anticonvulsive effects of lenalidomide (10 mg/kg) on the PTZ-induced clonic seizure thresholds in mice. Overall, these data raise the possibility of NO contribution to the anticonvulsive effects of lenalidomide in this animal model of seizure. Additionally, to assess the involvement of NMDAR pathway, we used the selective NMDAR antagonist MK-801 and the selective NMDAR agonist D-serine. Our data showed that pre-treatment with a per se non-effective dose of MK-801 was able to inhibit the anticonvulsive effects of lenalidomide. This finding also suggests a role for NMDAR in the lenalidomide’s effects on clonic seizure threshold. Based on the results of both NOS inhibitors and NMDAR antagonist in our study, the data can suggest the involvement of NMDAR and NO signaling in this effect of lenalidomide. It is well established that activation of NMDARs results in the activation of nNOS and related NO production and its downstream in the CNS.23–26 Accordingly, the results of the nNOS inhibitor 7-NI are in parallel with the NMDAR antagonist MK-801 in our study; thus, the NMDAR/nNOS could be a potential contributor to the lenalidomide effects on clonic seizure threshold. On the other hand, AG, which is a selective iNOS inhibitor (not a nNOS inhibitor), was also able to decrease the anticonvulsive effects of lenalidomide. AG is shown to be over 50-fold more effective at inhibiting the enzymatic activity of iNOS than endothelial or neuronal NOS.27 This suggests that the source of NO in participating in the effects of lenalidomide on clonic seizure threshold in our study could be from either NMDAR/nNOS or iNOS activation.
Lenalidomide has been introduced as a derivative of thalidomide since 2004. It has been shown that clinical response to lenalidomide is maintained for a longer period of time in comparison with thalidomide in myelofibrosis patients.28 Moreover, lenalidomide exerts fewer side effects and has lower risks of peripheral neuropathy compared to thalidomide.28,29 Thalidomide’s effects on brain electroencephalography has been reported in pregnant rodents, the electrical activity of the fetus brain increased with slow waves of high amplitude;30 however, after administration of this glutaminergic drug, electrophysiological recording of cat’s brain was increased in the periods of slow waves and rapid eye movements, which were changed during sleep.31 For the first time, the antiepileptic effect of thalidomide was proposed by Palencia et al.8 in PTZ-provoked clonic seizures in rats. The same anticonvulsive effect again was reported later by the same group in another epilepsy model of amygdaloid kindling in rats.32 Consistently, in our recent studies, we found that thalidomide exerted anticonvulsive properties in PTZ-induced clonic seizure threshold in mice.10,22 One of the main findings of the present study was that the maximum anti-convulsive effect of lenalidomide was observed at a relatively low dose of lenalidomide (10 mg/kg). It is worth mentioning that the two major side effects of thalidomide and lenalidomide are teratogenicity and neuropathy in patients whose daily consumption reaches above 200 mg for long periods.32
Lenalidomide also possesses anti-neuroinflammatory effects.33 Motor deficits reduction, dopaminergic fiber loss, microgliosis, and pro-inflammatory cytokine expression were elucidated as the mechanisms of lenalidomide effects on a mouse model of Parkinson’s disease.33 There are several case reports of anticonvulsive effects of thalidomide not only on inflammatory-induced epilepsy, known as Rasmussen encephalitis, but also on non-inflammatory refractory epileptic patients.6,32 Payandemehr at al.22 indicated that constitutive NO pathway mediates the anticonvulsive activity of thalidomide. Additionally, modulatory effects of thalidomide through iNOS signaling pathway were proposed in morphine dependency in mice.13 The role of nNOS in the thalidomide effects in immunomodulation has been reported in some studies.11,22 iNOS modulation has also been proposed as an underlying mechanism in immunomodulatory effects of lenalidomide.11 Furthermore, it was recently proposed that in mechanism of morphine dependency, lenalidomide could modulate NO level elevation in brain through activating phosphoinositide 3-kinase enzyme in brain, which leads to increasing cellular NO levels.13 Our current data are also in line with previous findings, which emphasizes a role for NO signaling in mediating the anticonvulsive effects of lenalidomide in mice.
The interaction of NO and NMDARs in the pathophysiology of seizure have been investigated.34,35 NMDARs are introduced as pivotal modulators of nNOS activity and NO release in the CNS.26,36 It was reported in clinical studies that felbamate, an NMDAR antagonist, demonstrated a therapeutic effect on controlling refractory partial seizures in humans.34,35 Although glutamate receptors are mainly known for their pro-convulsive properties, interaction of NMDA and NO in pathophysiology of both anticonvulsive and pro-convulsive effects of D-penicillamine on PTZ-induced clonic seizures in mice have been proposed.19 In studies evaluating the regulation of seizure susceptibility, it has been demonstrated that either NOS substrates or NO donors can exhibit both anticonvulsive37–39 or pro-convulsive40–43 effects in a variety of pre-clinical seizure models; this contradiction may be due to different experimental conditions (e.g., using different pharmacological tools to modify the NO signaling) in these studies. Accordingly, NO is demonstrated to contribute to both anticonvulsive and pro-convulsive effects of morphine on PTZ-induced clonic seizure in mice44,45 or lithium/pilocarpine-induced status epilepticus in rats.46 NOS inhibitors are also shown either to potentiate38,47–50 or to inhibit 42,47,50–52 experimentally induced seizures. This bimodal effects of NO on seizure susceptibility are in parallel with the fact that NO could exert either neuroprotective or neurotoxic effects in a variety of animal models of neurologic diseases.24,25 The levels of NO produced by different NOS isoforms could play an important role in this regard; at the lowest levels (usually nanomolar concentrations), normal physiologic role of NO is achieved primarily through reaction with ferrous hemes and related cyclic guanosine monophosphate-mediated signaling as well as with reactive radicals.53 However, excess levels of NO can interact with a variety of cytosolic, nonheme iron targets or convert into reactive nitrogen species leading to nitrosative, nitrative and oxidative modifications which are generally neurotoxic.23–25
Glutamate is the major excitatory neurotransmitter in the CNS and acts on ionotropic and metabotropic glutamate receptors, including NMDARs and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor, which regulate neuronal excitability and synaptic plasticity.54,55 NO physiologically regulates activity of these receptors via S-nitrosylation,54,56 which is a reversible, covalent addition of NO group to a cysteine thiol/sulfhydryl to form an S-nitrosothiol derivative.57 NO S-nitrosylates GluN2A subunit of NMDAR, which inhibits NMDAR activity.56 This effect affords a negative feedback system for glutamate neurotransmission preventing excitotoxicity.56 S-Nitrosylation of NMDAR may induce a conformational change in the receptor protein that makes glutamate and Zn2+ bind more tightly to the receptor, causing the receptor to desensitize and, consequently, the ion channel to close.58 Therefore, one may suggest that the inhibition of NOS could suppress this regulatory effect of NO on glutamatergic receptors and increase the susceptibility for excitotoxicity and seizure progression.50
The dose of the NMDAR antagonist MK-801 (0.05 mg/kg) that we currently used did not individually affect the seizure thresholds, which is in consistent with our previous study.17 We found that combining lenalidomide with this non-effective dose of MK-801 reversed anticonvulsive effects of lenalidomide. Previously, we showed that MK-801 at higher doses such as 1 mg/kg inhibits clonic seizure thresholds induced by PTZ in mice,17 which is in agreement with the overall anticonvulsive property of NMDAR antagonists in pre-clinical studies.59–61 However, paradoxical pro-convulsive effects of MK-801 on seizure threshold have been also reported.38,62 The mechanisms underlying such paradoxical effects of MK-801 on seizure threshold remain to be elucidated. One possible explanation could be the differences in the distribution of NMDARs in the brain, as activation of NMDARs in certain brain regions can inhibit seizures.38 For instance, it was reported that direct injection of the NMDAR agonist NMDA into the corpus striatum 63 or substantia nigra pars compacta64 acted to release gamma aminobutyric acid (GABA) and dopamine, respectively, and inhibited pilocarpine-induced seizures in rodents. Thus, inhibition of NMDARs in these brain regions may be pro-convulsive. Clearly, more studies are needed to explore the mechanisms underlying paradoxical effects of NMDARs on seizure threshold in different brain regions.
The anticonvulsive effects of thalidomide have recently been proposed in animal models and human’s studies. The majority of hypnotics and sedatives drugs have shown parallel anti-epileptogenic properties; thus, it seems logical that an analog of glutamic acid agent, thalidomide, and its derivative, lenalidomide, show the same anticonvulsive activity, as found in our present study. Of noted, there is no report of physical dependence to thalidomide or its derivative agent lenalidomide, in contrast with other hypnotic and sedative drugs. Moreover, in suicidal attempts by these drugs, survival even after huge doses consumption has been reported.65,66 In the current study we used PTZ (a GABA receptor antagonist) to create a common chemically-induced clonic seizures in mice. The PTZ-induced seizures are categorized as a model of generalized seizure that produces a myoclonic seizure.67 When screening anticonvulsant candidates the PTZ model is a good tool for evaluating anti-seizure characteristics compared to a focal or partial seizure model.67 Beside assessing the time until the first whole body clonus event, some studies also assess time to the tonic-hind limb extension after PTZ injection, which may extend assessment for tonic seizures.68 However, it is noteworthy that in our study we only assessed the clonic seizures (and did not evaluate the time to tonic phase) after PTZ injection. In conclusion, our data indicated that acute administration of lenalidomide increases the PTZ-induced clonic seizure threshold in mice. Additionally, we demonstrated that this anticonvulsive activity of lenalidomide may be mediated by the NMDAR and NO pathway.